Import RD signal for each sample from bam/sam/cram file and calculate histograms with specified bin size. It can be any positive integer divisible by 100. We use 100000 in this example
> cnvpytor -root sample.pytor -rd sample.bam
> cnvpytor -root sample.pytor -his 100000
Import BAF signal for each sample from vcf file and calculate histograms with specified bin size.
> cnvpytor -root sample.pytor -snp sample.vcf.gz
> cnvpytor -root sample.pytor -mask_snps
> cnvpytor -root sample.pytor -baf 100000
Second line is used to filter out all SNP-s that are not in P-region of the strict 1kG mask.
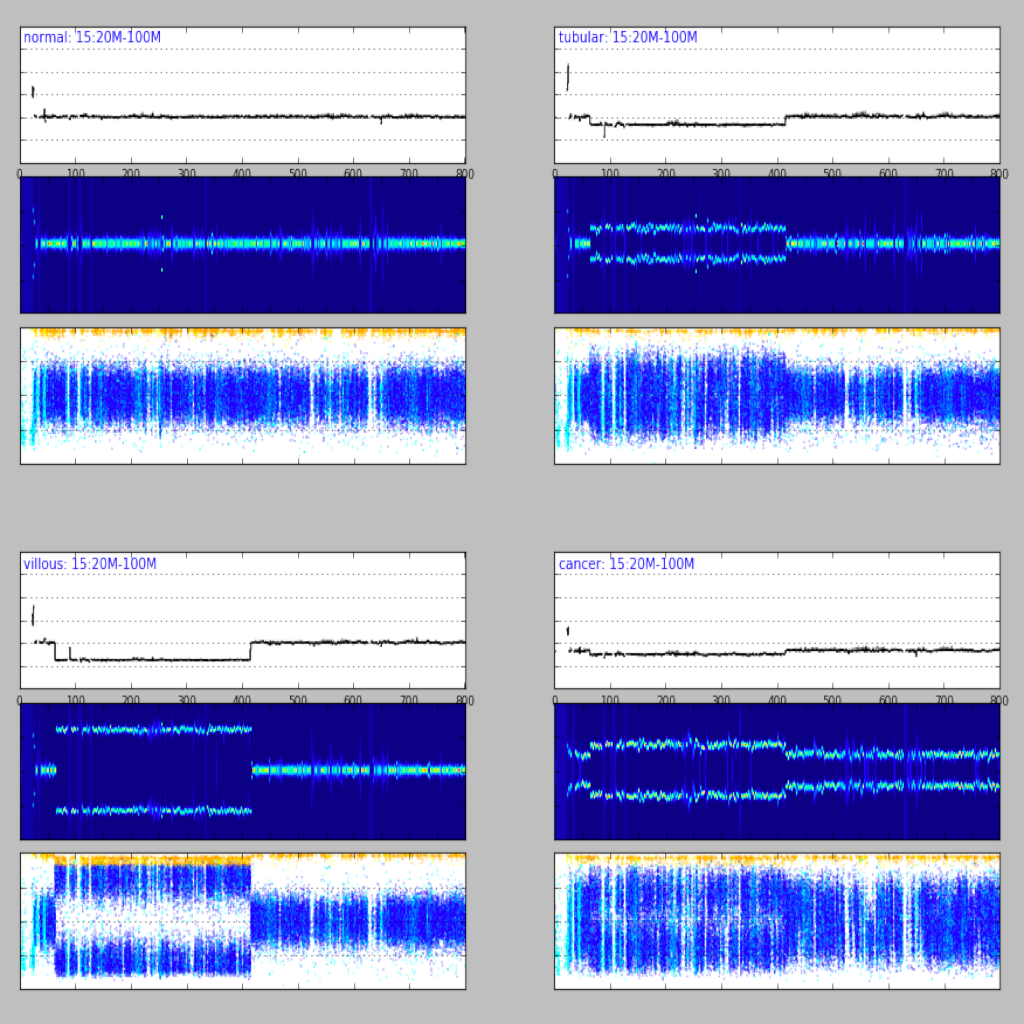
Enter interactive plotting mode with all sample you want to plot listed:
> cnvpytor -root sample1.pytor sample2.pytor sample3.pytor sample4.pytor -view 100000
cnvpytor> set style classic
cnvpytor> set rd_use_mask
cnvpytor> set file_titles normal tubular villous cancer
cnvpytor> set panels rd likelihood snp
cnvpytor> set markersize 0.2
cnvpytor> 15:20M-100M
cnvpytor> save image.png
cnvpytor> quit
By pressing tab two times after typing 'set style ' you will got list of all available styles.
If you skip third line (set file_titles) file names will be used for subplot title.
Instead 15:20M-100M you can specify any genomic region or regions. If there are more then one region they can be comma or space separated (comma - to be plotted in the same subplot, space - to be separately plotted).
Instead 'quit' CTRL+D can be used.
In this example we used data from following article:
[1] Oncotarget 2017 Dec 26;9(6):6780-6792. doi: 10.18632/oncotarget.23687
Inferring modes of evolution from colorectal cancer with residual polyp of origin.
Kim M, Druliner BR, Vasmatzis N, Bae T, Chia N, Abyzov A, Boardman LA.